Pymatgenによるバンド情報の取得
本記事の概要
この記事では、Pymatgenによるバンド情報取得方法を紹介していく。
今回は、バンド図(パスタみたいなグラフ)とDOS(Density od State)を描くところまで行う。
Pythonの基礎を習得していることを前提として解説を進めていく。
目次:
バンド図の表示¶
バンド情報を取得するためには、Material IDが必要になる。事前に対象材料のMatarial IDを取得しておく必要がある。
import matplotlib from pymatgen import MPRester from pymatgen.electronic_structure.plotter import BSPlotter mp_id='mp-3748' #AlCuO2 筆者が適当に選んだ化合物。 #api_key='YOUR_API_KEY' #自分でapi keyは取得してください。 def get_band(mp_id,api_key): with MPRester(api_key) as m: bs=m.get_bandstructure_by_material_id(mp_id) bg=bs.get_band_gap() dbg=bs.get_direct_band_gap() for k,v in zip(bg.keys(),bg.values()): print(k,':',v) print('direct_band_gap:',dbg) plotter=BSPlotter(bs) #plotter.get_plot().show() plotter.get_plot().savefig('BS_{}.png'.format(mp_id)) get_band(mp_id,api_key)
>(出力)
direct : False
energy : 1.8148
transition : (0.591,0.409,0.000)-\Gamma
direct_band_gap: 2.7100999999999997

筆者は、バンド図は詳しく読めないので解説は割愛する
DOSの表示
DOSを取得する場合も、同様にMaterial IDが必要となるので注意。
import matplotlib from pymatgen import MPRester from pymatgen.electronic_structure.plotter import BSPlotter,DosPlotter def get_dos(mp_id,api_key,xmin=None,xmax=None,ymin=None,ymax=None): with MPRester(api_key) as m: data = m.get_dos_by_material_id(mp_id) print(data) plotter=DosPlotter() name='{}_total_DOS'.format(mp_id) plotter.add_dos(name,data) plotter.get_plot(xlim=[xmin,xmax],ylim=[ymin,ymax]).savefig(name) get_dos(mp_id,api_key)
>(出力)
Complete DOS for Full Formula (Al1 Cu1 O2)
Reduced Formula: AlCuO2
abc : 5.931859 5.931859 5.931859
angles: 28.086128 28.086128 28.086126
Sites (4)
# SP a b c
-
-
- ---- -------- -------- --------
-
0 Al 0.5 0.5 0.5
1 Cu 0 0 0
2 O 0.889957 0.889957 0.889957
3 O 0.110043 0.110043 0.110043

まとめ
上記の方法で簡単にバンド情報を取得することができる。
ただしMaterials Projectに掲載されている第一原理計算結果の情報は、結構荒い精度の計算で出されているものが多いので精度の高さには注意が必要になってくるかもしれない。それでも網羅的にバンド情報を取得できるのはすごく便利である。
Xenonpyによる記述子の作成
本記事の概要
本記事では、Xenonpyを用いた記述子の作成方法を紹介する。
Pythonの基礎は習得していることを前提に話を進めていく。
目次:
Xenonpyとは
XenonPyは、統計数理研究所ものづくりデータ科学研究センターのチーム (Chang Liu 特任助教、野口瑶 特任研究員、Stephen Wu 助教、山田寛尚 特任研究員、吉田亮 同センター長) が物質・材料研究機構と共同開発しているマテリアルズインフォマティクス (MI) のオープンソースプラットフォーム。XenonPyには、様々なマテリアルインフォマティクスのアプリケーションのためのツールが豊富に用意されている。記述子生成クラスでは、組成、構造から数種類の数値記述子を計算することができる。
以下、公式ドキュメント
xenonpy.readthedocs.io
早速、実装
Xenonpyのインストール方法は、以下のサイトを参考にしてください。
xenonpy.readthedocs.io
Xenonpyを使用するために、以下のrdkitをインストールしておく必要がある。
! conda install --yes -c rdkit rdkit #最初にinstallしてください。
ライブラリのインポート
import pandas as pd import numpy as np import os, sys,warnings warnings.filterwarnings('ignore') from pymatgen.io.cif import CifParser from pymatgen.core.composition import Composition from xenonpy.descriptor import Compositions,Structures
組成記述子の作成
与えられた化学組成に対して290種類の組成特性を計算することができるらしい。
全ての記述子を確認したい方は、以下のURLをご確認ください。
xenonpy.readthedocs.io
XenonpyのCompositionsクラスでは、辞書形式に変換された組成から記述子を計算することができる。
注意)Compositionsは、Xenonpyの記述子を計算するためのクラス、CompositionはPymatgenの組成を辞書形式に変換してくれるクラス。自分で組成を辞書形式にするのが面倒であるため、PymatgenのCompositionクラスを使用している。 そして、Compositionsクラスに”composition”という列名があるデータフレームを渡すと組成情報から組成記述子を作成することができる。
def comp_descriptors(comp_holder): comp_holder = [Composition(x) for x in comp_holder] #Change strings(compositions) into dictionary df = pd.DataFrame({'composition':comp_holder}) cal_comp = Compositions() descriptor_comp = cal_comp.transform(df) #Make descriptors return descriptor_comp comp_holder = [ 'Li2O', 'MgO', 'LiCoO2', 'MgCo2O4', 'Fe' ] df = comp_descriptors(comp_holder) df
>(出力)

構造記述子の作成
構造記述子は、RDF(radial distribution function) OFM (orbital field matrix)2) を使用している。
参考)Pham et al. Machine learning reveals orbital interaction in materials, Sci Technol Adv Mater. 18(1): 756-765, 2017.
私が作成したプログラムではcifファイルが入っているディレクトリで、以下の関数を実行する必要がある。 そのためcifファイルが入っているディレクトリへのパスを引数で渡している。
Xenonpyでは、cifではなくPOSCARを読み取り構造記述子を作成する。この変換のプログラムなどは作成するのが面倒なのでPymatgenを使用している。そして、POSCAR形式の情報をデータフレームに格納し、その列名を"structure"にしておくことで、Structureクラスが読み取ることができ、構造記述子を計算することができる。
def getciflist(path_to_cif): ls = os.listdir(path_to_cif) calclist = [] for i in ls: if '.cif' in i: calclist.append(i.rstrip()) return calclist def str_descript(path_to_cif): #strはstructureの略 cif_list = getciflist(path_to_cif) str_holder = [] cif_holder = [] for cif in tqdm(cif_list): #時間がかかる場合があるので、tqmdでprogressバーを表示している cif_p = CifParser(cif)#cifの読み込み str_holder.append(cif_p.get_structures())#cifをPOSCARに変換している。 df = pd.DataFrame({'structure':str_holder}) cal_str = Structures() descriptor_str = cal_str.transform(df)#Make descriptors return descriptor_str cwd='./' df = str_descript(cwd) df
>(出力)

今回は、3つのcifファイルを用意したので、3つの結晶構造について記述子を獲得できている。
構造記述子は、合計で約1200個程度であることがわかる。
おまけ 組成記述子と構造記述子を同時に作成
実は、cifファイルがあれば、組成情報もcifファイルに書かれているので cifファイルから、組成記述子・構造記述子両方を作成することが可能である。 それが以下の関数になっている。
def getdescripts(path_to_cif): cif_list = getciflist(path_to_cif) str_holder = [] comp_holder =[] cif_holder = [] for cif in tqdm(cif_list):#時間がかかる場合があるので、tqmdでprogressバーを表示している with open(cif) as r: for i in r: if '_chemical_formula_sum' in i: comp_str = '' for j in i.split("\'")[1].split(): comp_str += j comp_holder.append(Composition(comp_str)) cif_p = CifParser(cif) str_holder.append(cif_p.get_structures()) cif_holder.append(cif) df = pd.DataFrame({'cif':cif_holder,'structure':str_holder,'composition':comp_holder}) df = df.set_index('cif') cal_comp = Compositions() cal_str = Structures() descriptor_comp = cal_comp.transform(df) descriptor_str = cal_str.transform(df) descriptor_comp_str = pd.merge(descriptor_comp,descriptor_str,left_index=True, right_index=True) return descriptor_comp_str cwd='./' df = getdescripts(cwd) df
>(出力)

組成記述子と構造記述子両方欲しい場合には、こんな感じにプログラムをかくと便利。
#コードが読みにくくて、大変申し訳ない。。。
Pymatgenによる化合物データの取得(Materials Project)
本記事では、Pymatgenによる化合物データ取得方法を紹介していく。
Pythonの基礎を習得していることを前提として話を進めていく。
目次:
Pymatgenとは
pymatgenとは、材料解析のためのオープンソースのpythonライブラリであり、以下の特徴があげられる。
原子や分子の構造をPythonのクラスで柔軟に表現
- VASPやGaussianといった多くのフォーマットに対応
- 位相図の作成や拡散解析などが可能な強力な解析ツール
- 状態密度やバンド構造など、電子構造解析も可能
- 結晶学のオープンデータベースであるMaterials Project REST APIと連動(今回はここ)
データベースにアクセスするためには、API KEYの取得が必要になる。
以下から、登録が可能である。
materialsproject.org
また、Pymatgenの公式ドキュメントは以下である。
pymatgen.org
元素情報の取得
まず、元素に関する情報を取得してみる。今回は、"Si"を例に紹介する。
元素情報は、非常に簡単に取得が可能である。
import pymatgen as mg el = mg.Element('Si') print(el.atomic_mass) #原子量を表示 print(el.melting_point) #融点を表示
>(出力)
28.0855 amu
1687.0 K
このように、非常に簡単に元素情報を取り出すことができる。
取り出せる全ての物性値を以下に示す(少し古い情報なので、今は変更があるかも)。
- Electrical resistivity
- Electronic structure
- Oxidation states
- Reflectivity
- Poissons ratio
- Molar volume
- Van der waals radiu
- ICSD oxidation states
- Bulk modulus
- Mineral hardness
- Mendeleev no
- Atomic no
- Boiling point
- Common oxidation states
- Brinell hardness
- Density of solid
- Thermal conductivity
- Critical temperature
- Name
- Atomic mass
- Coefficient of linear thermal expansion
- Rigidity modulus
- Youngs modulus
- Velocity of sound
- X
- Liquid range
- Atomic radius calculated
- Vickers hardness
- Superconduction temperature
- Melting point
- Refractive index
- Ionic radii
- Atomic radius
上記の物性を一つ一つ取ってくるのは、めんどうなので別の方法がある。
以下のように"el.data"とやると、辞書式で全ての物性値が取り出せる。
なので、for文を使って、全ての特性を簡単に取得することができる。
# 元素名を入れると、物性値が返ってくる関数を作成した。 def get_element_data(name): el = mg.Element(name) for i in el.data: print(i + ' : ' + str(el.data[i])) name='Er' get_element_data(name)
>(出力)
Atomic mass : 167.259
Atomic no : 68
Atomic orbitals : {'1s': -1961.799176, '2p': -302.01827, '2s': -316.310631, '3d': -51.682149, '3p': -63.818655, '3s': -70.310142, '4d': -6.127443, '4f': -0.278577, '4p': -10.819574, '4s': -13.423547, '5p': -0.935202, '5s': -1.616073, '6s': -0.134905}
Atomic radius : 1.75
Atomic radius calculated : 2.26
Boiling point : 3141 K
Brinell hardness : 814 MN m-2
Bulk modulus : 44 GPa
Coefficient of linear thermal expansion : 12.2 x10-6K-1
Common oxidation states : [3]
Critical temperature : no data K
Density of solid : 9066 kg m-3
Electrical resistivity : 86.0 10-8 Ω m
Electronic structure : [Xe].4f12.6s2
ICSD oxidation states : [3]
Ionic radii : {'3': 1.03}
Liquid range : 1371 K
Melting point : 1802 K
Mendeleev no : 22
Mineral hardness : no data
Molar volume : 18.46 cm3
Name : Erbium
Oxidation states : [3]
Poissons ratio : 0.24
Reflectivity : no data %
Refractive index : no data
Rigidity modulus : 28 GPa
Shannon radii : {'3': {'VI': {'': {'crystal_radius': 1.03, 'ionic_radius': 0.89}}, 'VII': {'': {'crystal_radius': 1.085, 'ionic_radius': 0.945}}, 'VIII': {'': {'crystal_radius': 1.144, 'ionic_radius': 1.004}}, 'IX': {'': {'crystal_radius': 1.202, 'ionic_radius': 1.062}}}}
Superconduction temperature : no data K
Thermal conductivity : 15 W m-1 K-1
Van der waals radius : no data
Velocity of sound : 2830 m s-1
Vickers hardness : 589 MN m-2
X : 1.24
Youngs modulus : 70 GPa
Metallic radius : 1.756
iupac_ordering : 36
IUPAC ordering : 36
化合物情報の取得
pymatgenでは、物性値の取得方法は2種類ある(私の知る限り)。
- 組成を入力する
- 条件を指定して習得する
まずは、組成情報を入力して取得する方をやってみる。
組成情報を入力して取得する
取得できる物性情報は、selected_propに書いてある物性値である。
早速、データを取得してみる。
import pymatgen as mg from pymatgen import MPRester #バージョンなどによって、MPResterの位置が異なる場合がある。 # その時は、pymatgenのライブラリを覗きに行ってMPResterの位置を確認にてあげてください。 selected_prop =[ "energy", "energy_per_atom", "volume", "energy", "formation_energy_per_atom", "nsites", "unit_cell_formula", "pretty_formula", "is_hubbard", "elements", "nelements", "e_above_hull", "hubbards", "is_compatible", "spacegroup", "task_ids", "band_gap", "density", "icsd_id", "icsd_ids", #"cif", #表示するとかなり大きくなってしまうので、今回は取得しない。 "total_magnetization", "material_id", "oxide_type", "tags", "elasticity" ] api_key='YOUR_API_KEY' #'SET YOUR API' #自分のAPI keyを入力してください。 with MPRester(api_key=api_key) as m: mater = 'ZnMnO3' #ここに欲しい化合物の組成を書く。 data = m.get_data(mater) #この行で、データを取得している。 for i in data: print('='*30) for j in selected_prop: print(str(j) + ' : ' + str(i[j]))
>(出力)
==============================
energy : -120.56779976
energy_per_atom : -6.028389988
volume : 195.097888591396
energy : -120.56779976
formation_energy_per_atom : -1.7328056347782081
nsites : 20
unit_cell_formula : {'Mn': 4.0, 'Zn': 4.0, 'O': 12.0}
pretty_formula : MnZnO3
is_hubbard : True
elements : ['Mn', 'O', 'Zn']
nelements : 3
e_above_hull : 0.09838204933333117
hubbards : {'Mn': 3.9, 'Zn': 0.0, 'O': 0.0}
is_compatible : True
spacegroup : {'symprec': 0.1, 'source': 'spglib', 'symbol': 'Pnma', 'number': 62, 'point_group': 'mmm', 'crystal_system': 'orthorhombic', 'hall': '-P 2ac 2n'}
task_ids : ['mp-900209', 'mp-772528', 'mp-885520', 'mvc-3995', 'mp-885529', 'mp-1274943', 'mp-1295178', 'mp-1314552', 'mp-1321473', 'mp-1317304', 'mp-1324033', 'mp-1638554', 'mp-1638625', 'mp-1638637', 'mp-1638619', 'mp-1651804', 'mp-1650749', 'mp-1787315', 'mp-900941', 'mp-1979901', 'mp-1981003', 'mp-1980086']
band_gap : 0.11529999999999996
density : 5.731355844333344
icsd_id : None
icsd_ids :
total_magnetization : 2.9994051
material_id : mp-772528
oxide_type : oxide
tags :
elasticity : None
==============================
energy : -27.82799765
energy_per_atom : -5.56559953
volume : 52.26198479368501
energy : -27.82799765
formation_energy_per_atom : -1.2700151767782075
nsites : 5
unit_cell_formula : {'Mn': 1.0, 'Zn': 1.0, 'O': 3.0}
pretty_formula : MnZnO3
is_hubbard : True
elements : ['Mn', 'Zn', 'O']
nelements : 3
e_above_hull : 0.5611725073333318
hubbards : {'Mn': 3.9, 'Zn': 0.0, 'O': 0.0}
is_compatible : True
spacegroup : {'symprec': 0.1, 'source': 'spglib', 'symbol': 'Pm-3m', 'number': 221, 'point_group': 'm-3m', 'crystal_system': 'cubic', 'hall': '-P 4 2 3'}
task_ids : ['mp-1016932', 'mp-1017012', 'mp-1017069', 'mp-1017050', 'mp-1430103', 'mp-1731940', 'mp-1794974', 'mp-1615980']
band_gap : 0.0
density : 5.348894748303294
icsd_id : None
icsd_ids :
total_magnetization : 3.1400626
material_id : mp-1016932
oxide_type : oxide
tags :
elasticity : {'G_Reuss': 61.0, 'G_VRH': 67.0, 'G_Voigt': 73.0, 'G_Voigt_Reuss_Hill': 67.0, 'K_Reuss': 76.0, 'K_VRH': 76.0, 'K_Voigt': 76.0, 'K_Voigt_Reuss_Hill': 76.0, 'elastic_anisotropy': 0.96, 'elastic_tensor': [[225.0, 1.0, 1.0, 0.0, 0.0, 0.0], [1.0, 225.0, 1.0, 0.0, 0.0, 0.0], [1.0, 1.0, 225.0, 0.0, 0.0, 0.0], [0.0, 0.0, 0.0, 47.0, 0.0, 0.0], [0.0, 0.0, 0.0, 0.0, 47.0, 0.0], [0.0, 0.0, 0.0, 0.0, 0.0, 47.0]], 'homogeneous_poisson': 0.16, 'poisson_ratio': 0.16, 'universal_anisotropy': 0.96, 'elastic_tensor_original': [[223.52508206920976, 2.624401788820888, 1.2895446005609252, 0.0, 0.0, 0.0], [0.21993485643860408, 225.78851072542224, 1.2971077031573202, 0.0, 0.0, 0.0], [0.21964388164024937, 2.6278009860233453, 224.5228546177142, 0.0, 0.0, 0.0], [-0.3447858513789858, 0.4339506851651542, -0.02077365489007688, 46.931401255089256, 0.0, 0.0], [-0.21077425914030695, 0.35241040047770134, -0.18308315075587742, 0.0, 46.92248801527998, 0.0], [0.018810794127094042, -0.038505358317629475, -0.007882863514756009, 0.0, 0.0, 46.932124201983065]], 'compliance_tensor': [[4.5, -0.0, -0.0, 0.0, -0.0, 0.0], [-0.0, 4.5, -0.0, 0.0, 0.0, 0.0], [-0.0, -0.0, 4.5, -0.0, -0.0, 0.0], [0.0, -0.0, -0.0, 21.3, -0.0, 0.0], [-0.0, 0.0, -0.0, 0.0, 21.3, -0.0], [0.0, 0.0, 0.0, -0.0, -0.0, 21.3]], 'warnings': , 'nsites': 5}
==============================
energy : -61.20978206
energy_per_atom : -6.120978206
volume : 102.91461477749
energy : -61.20978206
formation_energy_per_atom : -1.8253938527782083
nsites : 10
unit_cell_formula : {'Mn': 2.0, 'Zn': 2.0, 'O': 6.0}
pretty_formula : MnZnO3
is_hubbard : True
elements : ['Mn', 'O', 'Zn']
nelements : 3
e_above_hull : 0.005793831333330779
hubbards : {'Mn': 3.9, 'Zn': 0.0, 'O': 0.0}
is_compatible : True
spacegroup : {'symprec': 0.1, 'source': 'spglib', 'symbol': 'R-3', 'number': 148, 'point_group': '-3', 'crystal_system': 'trigonal', 'hall': '-R 3'}
task_ids : ['mp-770583', 'mp-803358', 'mp-811615', 'mp-754318', 'mp-830632', 'mp-1298159', 'mp-1292723', 'mp-1284960', 'mp-1276065', 'mp-1293892', 'mp-1298358', 'mp-1297282', 'mp-1296202', 'mp-1298447', 'mp-1296247', 'mp-1286252', 'mp-1286337', 'mp-1281904', 'mp-1291948', 'mp-1295441', 'mp-1279918', 'mp-1387979', 'mp-1752477', 'mp-1773640', 'mp-831650', 'mp-812278', 'mp-1621284']
band_gap : 1.7281
density : 5.432539520324601
icsd_id : None
icsd_ids :
total_magnetization : 2.99992435
material_id : mp-754318
oxide_type : oxide
tags : []
elasticity : None
こんな感じでデータが取得できる。
複数の化合物についてデータが取得されているが、これは入力した組成に多形が存在していることを意味している。
条件を指定して、取得する方法
含む元素を指定して化合物データを抽出する
La-Li-O-Zr4元系の化合物を取得のデータを取得してみる。
import pandas as pd #Material IDを取得して、テキストファイルに書き込む関数。 #この関数では、元素系を指定してMaterial IDを取得してきている。 def get_mp_ids(element_list,criteria,properties,output): properties=list(set(properties)) df=pd.DataFrame(columns=properties) #取得したデータをcsvにまとめたいため、pandasを利用する。 with open(output,'w') as w: with MPRester(api_key) as m: for i in [element_list]: data = m.query(criteria=criteria,properties=properties) #ここで条件と取得したい特性を指定している for d in data: #print(d) df.loc[d['material_id']]=list(d.values()) return df properties=[ "material_id", #"energy_per_atom", #"volume", #"energy", "formation_energy_per_atom", #"nsites", #"unit_cell_formula", "pretty_formula", #"is_hubbard", #"elements", #"nelements", "e_above_hull", #"hubbards", #"spacegroup", #"task_ids", "band_gap", #"density", #"icsd_id", #"icsd_ids", #"cif", #"total_magnetization", #"oxide_type", #"tags", #"elasticity" ] output = ':La-Li-O-Zr.txt' element_list = ['La','Li','O','Zr'] criteria={"elements": {'$in':[['La','Li','O','Zr' ]]}} #La-Li-O-Zr系の化合物を取得 #['La','Li','O','Zr' ]にすると、La,Li,O,Zrそれぞれを含む化合物(7万件くらい)をとってくるので注意 # 書き方の参考: https://pymatgen.org/pymatgen.ext.matproj.html df = get_mp_ids(element_list,criteria,properties,output) df

また、criteriaを"*-Li-O-Zr"とすると、Li-O-Zrを同時に含む全ての4元系化合物を取得することができる。
criteria="*-Li-O-Zr" #Li-O-Zrを同時に含む化合物を全て取得する df = get_mp_ids(element_list,criteria,properties,output) df

例:Li-O系化合物の取得
criteria={"elements": {"$all": ['Li','O']}}とすることで、LiとOを含む全ての化合物を取得することができる。
properties=[
"material_id",
#"energy_per_atom",
#"volume",
#"energy",
"formation_energy_per_atom",
#"nsites",
#"unit_cell_formula",
"pretty_formula",
#"is_hubbard",
#"elements",
#"nelements",
"e_above_hull",
#"hubbards",
#"spacegroup",
#"task_ids",
"band_gap",
#"density",
#"icsd_id",
#"icsd_ids",
#"cif",
#"total_magnetization",
#"oxide_type",
#"tags",
#"elasticity"
]
output = 'Li-O.txt'
criteria={"elements": {"$all": ['Li','O']}} #Liと0を同時に含む全ての化合物を取得する
df = get_mp_ids(element_list,criteria,properties,output)
df
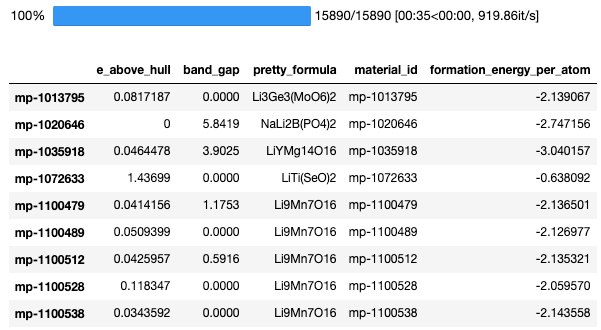
物性値で条件指定
criteria={"elements": {"$all": ['Li','O']},"band_gap":{"$gt":1}}とすると、 LiとOを同時に含むかつ、バンドギャップが1eV以上である化合物を取得することができる。
もちろん、他の物性値でも条件指定は可能である。
また、gtはgreater thanの意味である。 これらの数値条件の指定方法を下の表に示す。

output = 'Li-O_bandgap_gt1.txt' criteria={"elements": {"$all": ['Li','O']},"band_gap":{"$gt":1}} #バンドギャップが1eV以上であり、かつLiと0を同時に含む全ての化合物を取得 df = get_mp_ids(element_list,criteria,properties,output) df

まとめ
Pymatgenを使いこなすことができれば、このような巨大なデータベースから簡単にデータ取得ができるようになる。 また、化合物の解析も簡単に行えるようになる。 また、時間があるときに、Pymatgenの使い方を書いていこうと思う。
マテリアルズ・インフォマティクスにおけるデータ分析
本記事の概要
この記事では、マテリアルズ・インフォマティクスに限った話ではないが、機械学習を行う前のデータ分析について紹介していく。
グラフは発表などでも使えるように綺麗に作成している(論文でも使える?)ので、参考までにどうぞ。
目次:
データ分析の重要性
最近は機械学習のライブラリが発展しているため、すぐに機械学習にデータをかけて予測することができる。しかし、機械学習にかける前にデータ分析をすることで良い高い予測精度への改善ができたり、新しい相関関係などを見つけることができる。今回はそのデータ分析の実装方法をテンプレート的に紹介する。今回は、回帰予測を行うことを前提にデータ分析を行っている
データの準備
ここで扱うデータは前記事と同じくLi-O系の化合物に関する生成エネルギーに関するものである。データ分析を行うために、事前に以下を実行しておく。以下のコードは、ライブラリのインポート、データの読み込みである。
# データ加工・処理・分析ライブラリ import pandas as pd import numpy as np # 可視化ライブラリ import matplotlib.pyplot as plt import seaborn as sns %matplotlib inline #Li-O系の化合物データに関する記述子、目的変数が入っているファイル名 filename='./Li_O_atom2vec_ave_var_with_form_ene_per_atom.csv' df = pd.read_csv(filename,index_col=0) df.head()
目的変数に対する分析
今回は、目的変数に関して4つのデータ分析を行う。データが手に入ったら、まずやってみると良い(私は、大体この4つはやっている)。
ヒストグラムの作成
まずは、目的変数に対するヒストグラムから確認する。
#ここでは、ヒストグラムを描くための関数(サブルーティン)を作成している。 def make_hist(df,column_name,bins=100): #図を綺麗に書く設定 plt.rcParams['font.family'] = 'Times New Roman' #Font plt.rcParams['xtick.direction'] = 'in' # x axis in plt.rcParams['ytick.direction'] = 'in' # y axis in plt.rcParams['axes.linewidth'] = 1.0 # axis line width fig = plt.figure(figsize=(6,4),dpi=100) #FIgure Size ax = fig.add_subplot(111) #Figure position output_fig = 'out.{}.png'.format(column_name) #Image name ax.hist(df[column_name],bins=bins,color='k') ax.yaxis.set_ticks_position('both') ax.xaxis.set_ticks_position('both') ax.set_title('{} histogram'.format(column_name)) ax.set_xlabel(column_name) ax.set_ylabel('Frequency') plt.savefig(output_fig) print('Mean :',round(df[column_name].mean(),4)) print('Median :',round(df[column_name].median(),4)) print('output figure:',output_fig) plt.show() column_name='formation_energy_per_atom' make_hist(df,column_name)
>(出力)
Mean : -2.2949
Median : -2.3526
output figure: out.formation_energy_per_atom.png
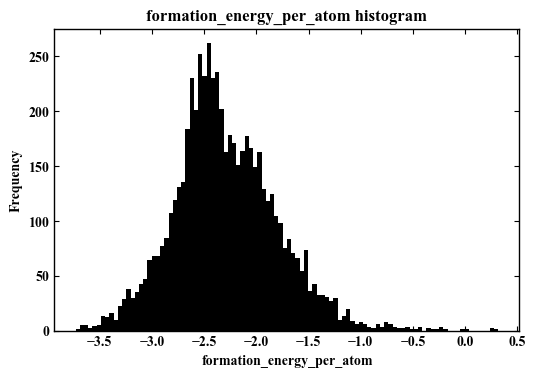
今回の目的変数は正規分布に近い形になっていることがわかる。よって、ある程度回帰問題として解きやすいだろうと推測できる。
データが明らかに2分しているような分布だったり、偏りが非常に大きい場合はうまく回帰できない場合が多い。そのような場合は、回帰問題ではなく、分類問題に変換した方が良いかもしれない。
箱ひげ図を描く
みんながよく知っている箱ひげ図
#ここでは、箱ひげ図(Boxplot)を描くための関数(サブルーティン)を作成している。 def make_box_plot(df,column_name): plt.rcParams['font.family'] = 'Times New Roman' plt.rcParams['xtick.direction'] = 'in' # x axis in plt.rcParams['ytick.direction'] = 'in' # y axis in plt.rcParams['axes.linewidth'] = 1.0 # axis line width fig = plt.figure(figsize=(6,4)) ax = fig.add_subplot(111) ax.boxplot(df[column_name]) ax.set_title('Boxplot plot {}'.format(column_name)) ax.set_ylabel('Count') ax.set_xlabel(column_name) ax.yaxis.set_ticks_position('both') print(df[column_name].describe()) column_name='formation_energy_per_atom' make_box_plot(df,column_name)
> (出力)
count 6033.000000
mean -2.294868
std 0.493983
min -3.732763
25% -2.610536
50% -2.352607
75% -1.989622
max 0.316142
Name: formation_energy_per_atom, dtype: float64
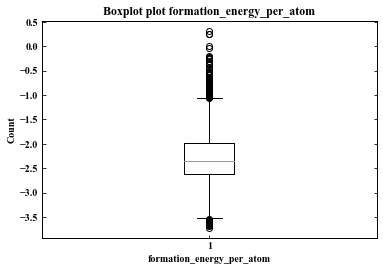
ここでは、明らかな外れ値があるかどうかを調べている。今回は、ひどい外れ値は見当たらないと考えられる。
機器トラブルなどで桁違いに外れている値であったり、物理的におかしい値がある場合はそれらのデータを除外してから機械学習にかけた方がよいだろう。
目的変数と記述子の相関関係
目的変数と記述子の相関係数を確認する。
def feature_plot(df_corr,column_name,labelsize=18): plt.rcParams['font.family'] = 'Times New Roman' plt.rcParams['xtick.direction'] = 'in' # x axis in plt.rcParams['ytick.direction'] = 'in' # y axis in plt.rcParams['axes.linewidth'] = 1.0 # axis line width fig = plt.figure(figsize=(14,6)) ax = fig.add_subplot(111) ax.bar(df_corr.index,df_corr['formation_energy_per_atom'], color='Black', linestyle=':') ax.set_ylabel('Correlation coefficient between target and descriptors',fontsize=labelsize) ax.set_xlabel('Features',fontsize=labelsize) ax.yaxis.set_ticks_position('both') ax.set_xticklabels(df_corr.index,rotation=90,fontsize=labelsize) ax.set_title('Correlation between target and descriptors',fontsize=labelsize*1.2) plt.show() column_name='formation_energy_per_atom' df_corr = df.corr().iloc[:-1] feature_plot(df_corr,column_name)
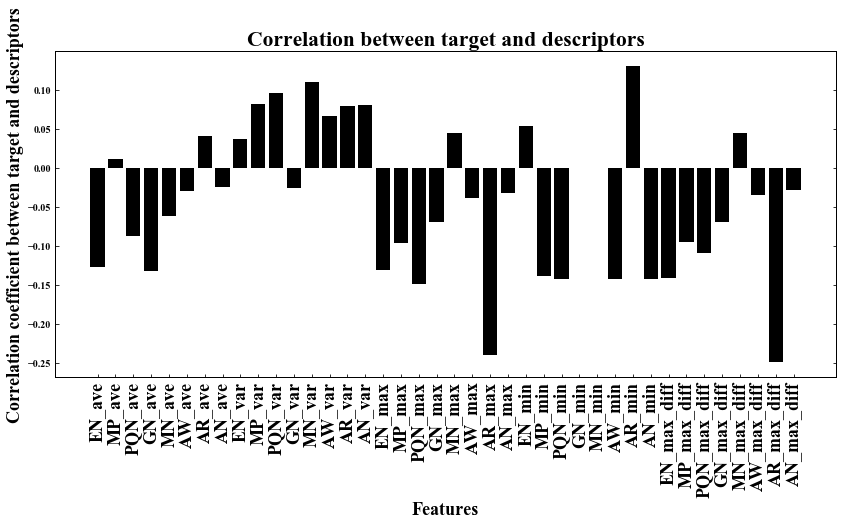
AR_max_diffが目的変数に対して、比較的大きな負の相関を持っていることがわかる。次に具体的にその相関状況を見てみる。
効きそうな記述子と目的変数の関係をさらに具体的に表示する
全ての記述子と目的変数の散布図を作成するのは、めんどくさいし、確認するにも時間がかかる。今回は、上の分析で一番相関係数の絶対値が大きかった記述子と目的変数の関係性をみる。なお、相関係数は、一時的相関しか反映されないため、二次的相関などそのほかの相関がある可能性もあるということを頭のすみに入れておいて方が良い。
def r(x,y): x_ = x.mean() y_ = y.mean() return (sum((x-x_)*(y-y_)))/ (np.sqrt((np.square(x-x_).sum()*(np.square(y-y_).sum())))) def plot(df,column1,column2,pos_x=0.9,pos_y=0.5): plt.rcParams['font.family'] = 'Times New Roman' plt.rcParams['xtick.direction'] = 'in' # x axis in plt.rcParams['ytick.direction'] = 'in' # y axis in plt.rcParams['axes.linewidth'] = 1.0 # axis line width x_max_value,y_max_value = max(df[column1]),max(df[column2]) fig = plt.figure(figsize=(12,8),dpi=100) ax = fig.add_subplot(221) ax.scatter(df[column1],df[column2],color='k',s=5) ax.set_xlabel(column1) ax.set_ylabel(column2) s='r = '+str(round(r(df[column1],df[column2]),4)) ax.text(x = x_max_value*pos_x, y = y_max_value*pos_y ,s=s) ax.yaxis.set_ticks_position('both') ax.xaxis.set_ticks_position('both') ax.set_title('{} vs {}'.format(column1,column2)) plt.show() column1='AR_max_diff' column2='formation_energy_per_atom' plot(df,column1,column2)

このグラフからは、それほど明らかな相関があるような形にはなっていないことが確認できる。
なお、上記の関数とFor文をうまく用いれば、全ての記述子と目的変数の散布図を簡単に作成することができる。
記述子間の相関分析
記述子間の関係性も重要である。多重共線性などを確認するのにも役立つため、ぜひ確認しておいて欲しい。 記述子間の相関を全て確認するのは、非常に手間がかかりそうだが、以下のプログラムを用いることで全ての記述子間の相関をヒートマップ的に確認することができる。非常に便利なので重宝している。
plt.rcParams['font.family'] = 'Times New Roman' plt.rcParams['xtick.direction'] = 'in' # x axis in plt.rcParams['ytick.direction'] = 'in' # y axis in plt.rcParams['axes.linewidth'] = 1.0 # axis line width fig = plt.figure(figsize=(12,12)) sns.heatmap(df.corr(),annot=False,cmap='bwr',linewidths=0.2) fig=plt.gcf() fig.set_size_inches(10,8) plt.show()
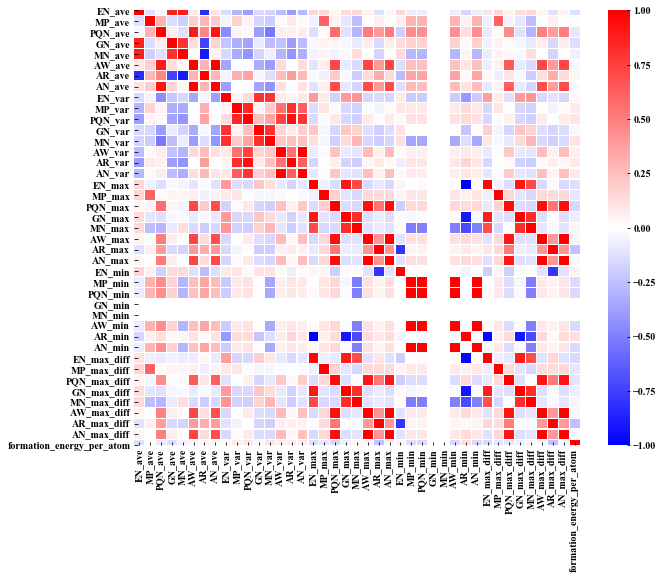
今回は、Li-O系の化合物についてのデータになっているため、GN_min(族番号の最小値)は、必ず1(Li)になり、また MN_min(メンデレーエフ番号の最小値)も必ず1(Li)になるため、相関は0となっている(白色)。
また、記述子間で相関係数が高い(0.5以上)のものがいくつかあるため、多重共線性が発生してしまうと考えられる。
ちなみに、変数同士の相関係数だけを確認したい場合は、以下のようにcorr()を使用すると相関係数が取得できる。
df_corr = df.corr() df_corr

まとめ
機械学習にかける前に、このような最低限のデータ分析を行うことが大切である。データ分析によって、新たな発見が、精度向上や知見を産むことが多々ある。また、異常値や物理的におかしい値などを発見することもできるため、それらを事前にすることも可能である。このようなテンプレートでも良いため、まずはデータ分析を行おう。
マテリアルズ・インフォマティクスにおける回帰係数・変数重要度の分析
本記事の概要
この記事では、機械学習を行った際の変数重要度・回帰係数の分析について解説していく。理論系ではなく、実装を主に書いていく。言語はPython、ライブラリはscikit-learnを使用している。pythonの基礎を習得した方向けの内容となっている。”マテリアルズ・インフォマティクスにおける〜〜”と言う記事題名にしておきながら、この辺の方法はマテリアルズ・インフォマティクスに限ったものではなく、機械学習一般における実装になる。
記述子の解析
線形系の予想モデルの回帰係数や決定木系のモデルの変数重要度を確認することが可能である。これによって、重要な変数だけを抽出して次の回帰の参考にすることも可能であるし、どのような変数が聞いているのか推測することもできる。マテリアルズ・インフォマティクスでは主に後者の方が重要かもしれない。とりあえず、今回は回帰係数・変数重要度の確認の仕方を解説していく。
回帰係数
回帰係数による解析は、非常に簡単であり、イメージつきやすいのでそれほど説明は不要であると思う。 回帰係数の絶対値が大きければ、重要な変数であることは間違いないし、変数の値が正になっていれば目的変数と正の相関があるなどといった具合である。
この回帰係数で変数の分析を行う場合は、スケーリングを事前に施した記述子を使用する必要があるという点に注意が必要である。これは、大きなスケールで変動する記述子と小さなスケールで変動する記述子では、回帰係数のスケールも異なってくるためである。
決定木の変数重要度とは何か
決定木の場合は、変数重要度を表す指標はいくつか存在する。しかし、gini不純度を用いた重要度が最も一般的であると言える。
gini不純度とは
gini不純度とは、ノードごとにラベルをどのくらい分類できていないかを示す指標となっている。あるノードに関するgini不純度は、以下の式で定義することができる。

上の式から、例えば、ノードにおいて完全にサンプルが分類されている場合は、gini不純度は0になることがわかるだろう。
変数の重要度
gini不純度を下に変数重要度を計算することができる。もう少し具体的に言うと、重要度は、ある特徴量で分割するとどれくらいgini不純度を下げられるのかを意味している。ある特徴量j に関する重要度は以下で定義することができる。

これらの説明は、以下のサイトがわかりやすいと思ったのでurlを載せておく。
yolo-kiyoshi.com
多重共線性
ただし、これら変数の分析を行う際には、変数同士の多重共線性に注意する必要がある。多重共線性とは、記述子同士の相関係数が高い場合に起こってしまう現象のことをいう。記述子どうし相関係数が高いと回帰係数や変数重要度が全く安定しないという現象が起きてしまう。つまり、回帰を行うたびに全く異なる係数・重要度が獲得されてしまい、その回帰係数・重要度は信頼できないものとなってしまう。 このような対処方法としては、事前に記述子同士の相関係数を確認し、そのような記述子を除外するなどの対処が必要となってくる。わかりやすい解説を行っているサイトのurlを以下に載せておく。
xica.net
toukeier.hatenablog.com
note.com
実装
モデル作成のための準備
モデルを作るために、ライブラリのインポート、データの読み込みが必要なのでいつもの通りそれらの準備をする。使用するデータは、Materials Project中のLi-O系化合物約6000件に関するものである。
今回は、目的変数を生成エネルギー、記述子を元素特性の統計量(約40個)を使用している。
詳しくは、前の記事を参考にしてください。
taruto-mateinfo.hatenablog.com
# データ加工・処理・分析ライブラリ import numpy as np import pandas as pd # 可視化ライブラリ import matplotlib.pyplot as plt %matplotlib inline from sklearn.metrics import mean_squared_error from sklearn.model_selection import train_test_split #RMSEを算出するための関数を定義 def rmse(y_true,y_pred): return np.sqrt(mean_squared_error(y_true,y_pred)) #変数の重要度をプロットするための関数 def feature_plot(df_coef,labelsize=18): plt.figure(figsize=(14,6)) plt.bar(df_coef['features'],df_coef['coef'], label=model.__class__.__name__, color='Black', linestyle=':') plt.ylabel('Feature importance (coefficient)',fontsize=labelsize) plt.xlabel('Features',fontsize=labelsize) plt.xticks(rotation=90) plt.tick_params(labelsize=labelsize) plt.title('Coefficient analysis ({})'.format(model.__class__.__name__),fontsize=labelsize*1.2) plt.show() #Li-O系の化合物データに関する記述子、目的変数が入っているファイル名 filename='./Li_O_atom2vec_ave_var_with_form_ene_per_atom.csv' df = pd.read_csv(filename,index_col=0) X = df.drop(['mp-id','formation_energy_per_atom'],axis=1) y = df['formation_energy_per_atom'] X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=777)
線形回帰の係数分析
まずは、線形回帰の変数の分析である。回帰係数を分析するためには予測式を作成しなければならないので最初にフィッティングを行う。 その後、回帰係数の分析を行っている。
from sklearn.linear_model import Ridge #https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.Ridge.html # スケーリング(標準化) from sklearn.preprocessing import StandardScaler sc = StandardScaler() sc.fit(X_train) X_train_std = sc.transform(X_train) X_test_std = sc.transform(X_test) model = Ridge(alpha=1.0) model.fit(X_train_std, y_train) y_train_pred = model.predict(X_train_std) y_test_pred = model.predict(X_test_std) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) coef=model.coef_#回帰係数の取得 features_columns=X.columns df_coef = pd.DataFrame({'features':features_columns,'coef':coef}) df_coef=df_coef.sort_values('coef',ascending=False) df_coef.to_csv('{}_coef.csv'.format(model.__class__.__name__))#回帰係数をcsvファイルに書き出している feature_plot(df_coef)
>RMSE(train):0.345 RMSE(test):0.340(出力)

AN(元素番号)の平均値が正に効いていることがよくわかる。またAW(原子量)とPQN(周期番号)の平均が大きく負に効いていることもわかった。このようにして回帰係数を分析することが可能である。
ただし、これらの変数間で相関関係があるものが含まれていると予想される(例えば、原子番号と原子量は同じような変動をするので、間違いなく相関がある)。したがって、多重共線性があると考えられる。よって、本気で分析をしたい場合は、事前に変数同士の相関関係を調べる、 Lasso, PLSなどを用いて変数を削減しておく必要があったり、検定を行ったりする必要もある(ここまでちゃんとやっているマテリアルズ・インフォマティクスの研究は少ないが)。
基本的に、線形モデル(Lasso, 重回帰)であれば同じような方法"model.coef_"で回帰係数を取り出すことができる。
Random Forestによる変数重要度
from sklearn.ensemble import RandomForestRegressor model = RandomForestRegressor(max_depth=15,n_estimators=500, random_state=777) model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) coef = model.feature_importances_#変数重要度の取得 features_columns=X.columns df_coef = pd.DataFrame({'features':features_columns,'coef':coef}) df_coef=df_coef.sort_values('coef',ascending=False) df_coef.to_csv('{}_coef.csv'.format(model.__class__.__name__)) feature_plot(df_coef)>RMSE(train):0.088 RMSE(test):0.211(出力)
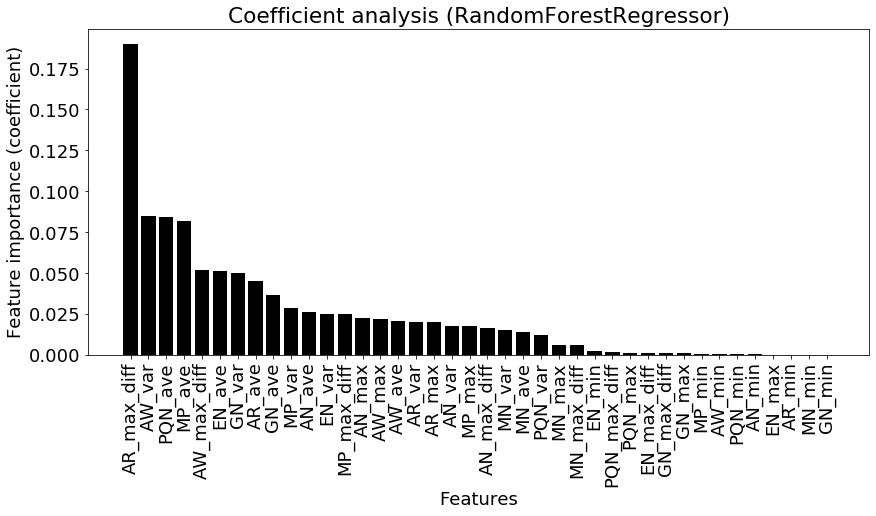
このscikit-learnを用いた場合は、gini不純度を元に変数重要度を算出している。
今回の分析で元素半径の最大差が最も重要度が高いことがわかる。次いで、原子量の平均値、周期番号の平均値が効いていると示されている。このように決定木のアンサンブルを使用したランダムフォレストは、精度も高くある程度変数の重要度を把握できるため、非常に多くの場面で使用されている。
ただし、線形回帰の回帰係数と同様に、この変数重要度に関しても多重共線性の影響が含まれているため、注意が必要である。
まとめ
scikit-learnを用いれば簡単に回帰係数や変数重要度を取得することができる。これらから、どのような要素が物性に効いているのかを調査したり、記述子の選定に使用したりすることが可能である。 ただし、多重共線性などに注意を払う必要があることを忘れずに。マテリアルズ・インフォマティクスのモデル評価とハイパーパラメータチューニング
本記事の概要
この記事では、以下について解説していく。
- モデルの評価方法(保留法と交差検証法)
- ハイパーパラメータチューニングの方法(グリッドサーチ)
本記事ではマテリアルズインフォマティクスで最もよく使用される教師あり学習のモデル評価方法とハイパーパラメータチューニングについて具体的なコードを交えて紹介していく。言語はPythonライブラリは、Scikit-learnを使用する。
この基礎は、Pythonをある程度習得した人向けの記事となっている。
目次:
モデルの評価方法
前回の記事では、学習データとテストデータに分けてモデルを作成し、評価を行った。
このようにモデルの学習に使用しないデータを予め準備し、モデルの性能を確認することは非常に重要である。これは、機械学習モデルは既存のデータに対して高い精度を達成するだけではなく、未知データに対しても高い性能を発揮することが重要となるからである。したがって、この章では、適切なモデル評価方法を学ぶこととする。
過学習とは:
ここで、機械学習でよく問題に挙げられる、過学習について説明する。機械学習では、学習に用いたデータに対しては高い精度が達成できるが、未知のデータに対しては精度が劣ってしまう現象を過学習という。機械学習は、基本的に過学習との戦いであると言われているくらいである。これを防ぐために、今回紹介する保留法や交差検証法を用いて評価を行う必要がある。
保留法と交差検証法
この節では、最もよく使用される評価手法である保留法と交差検証法を学ぶ。
保留法
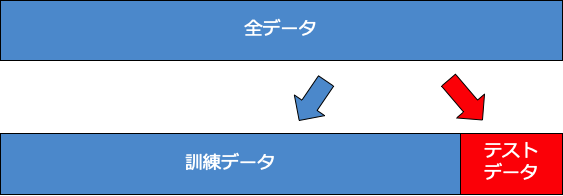
保留法(ホールドアウト法)とは、前回の記事でも行った通り、データを学習データとテストデータの二つにランダムに分割し、学習データを用いてモデルを作成してテストデータでモデルを評価する方法である。
保留法は、非常にシンプルな評価方法であるが、データ数が十分に大きい場合は、モデルの評価方法として十分に機能する。しかし、データ数が非常に少ない場合は、テストデータに偶然都合の良いデータが多くなってしまい、モデルが課題評価されてしまうという問題が生じる。下に保留法のイメージを示す。
交差検証法
交差検証法(Cross-validation)とは、上記の保留法の問題を解決できる手法である。これは、学習データと検証データを何度も入れ変えて評価する方法である。交差検証法で最もよく衣装っされるのがk分割交差検証法(K-fold cross-validation)である。この手法では、データをk個のブロックにランダムに分割し、k個のうち1つのブロックを検証データ、残りのk-1個を学習データとして学習する。 k分割交差検証法のイメージを下に示す。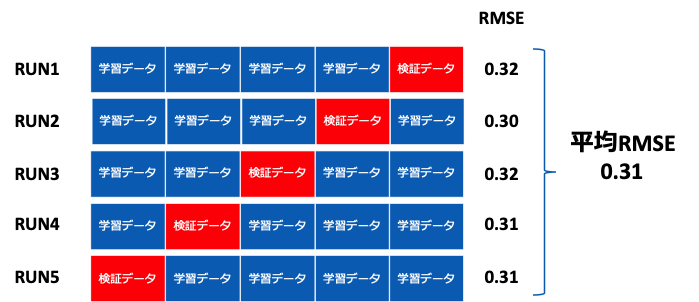
k分割交差検証法の実装準備
ここでは、Random Forestを用いて実際にk分割交差検証法を行う。扱うデータは前記事と同じくLi-O系の化合物に関する生成エネルギーの予測である。これを行うために、事前に以下を実行しておく。# 実装前の準備 # データ加工・処理・分析ライブラリ import numpy as np import pandas as pd # 可視化ライブラリ import matplotlib.pyplot as plt %matplotlib inline import sklearn # 機械学習ライブラリ import pickle # 予測モデルの保存をするためのライブラリ from sklearn.metrics import mean_squared_error #RMSEを算出するための関数を定義 def rmse(y_true,y_pred): return np.sqrt(mean_squared_error(y_true,y_pred)) #MAEを算出するための関数を定義(今回は使用しないがテンプレートとして残す) def mae(y_true,y_pred): return np.abs(y_true-y_pred).mean() #予測値と真値を比較するためのグラフを作成する関数を定義 def plot(y_true,y_pred): xx = np.arange(-4.0, 0.5, 0.1) plt.plot(xx,xx,linestyle='--') plt.scatter(y_true,y_pred) plt.xlabel('True values') plt.ylabel('Predicted values') plt.show() #Li-O系の化合物データに関する記述子、目的変数が入っているファイル名 filename='./Li_O_atom2vec_ave_var_with_form_ene_per_atom.csv' df = pd.read_csv(filename,index_col=0) #保留法による訓練データとテストデータの作成 from sklearn.model_selection import train_test_split X = df.drop(['mp-id','formation_energy_per_atom'],axis=1) y = df['formation_energy_per_atom'] X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=777)
k分割交差検証法の実装
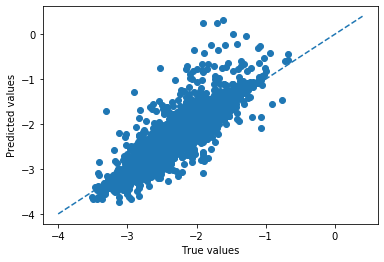
今回は、sklearn.model_selection.KFoldを用いてk分割交差検証法を行う。 n_split=5とすると、訓練データに対して5分割でk分割交差検証法を行うことになる。"for train_index,test_index in kf.split(X_train):" とすることで、ランダムに訓練データ用のインデックスと、検証データ用のインデックスが選択される。 そして、5分割の場合それが5回繰り替えされる。 このような交差検証を行うと、全てのトレーニングデータが必ず1度検証データになる。したがって、検証データになったときの予測値を集めていくことでデータ全体を使ってより検証精度を算出できる。このため、小さいデータセットであっても検証の信頼性が増すと言える。 また検証データに対する予測値のプロットも作ることができる。
from sklearn.ensemble import RandomForestRegressor #Ref.) https://scikit-learn.org/stable/modules/generated/sklearn.ensemble.RandomForestRegressor.html from sklearn.model_selection import KFold kf = KFold(n_splits=5,shuffle=True,random_state=777)#k分割交差検証法用のインスタンスを作成 pred_validation = np.array([])# 検証データになった際の予測値を集めるためのnp.array ans_validation = np.array([])# 検証データになった際の目的変数を順番通りに集めるためのnp.array pred_train = np.array([])# 訓練データになった際の予測値を集めるためのnp.array ans_train = np.array([])# 訓練データになった際の目的変数を順番通りに集めるためのnp.array for train_index,test_index in kf.split(X_train): X_tr = X_train.iloc[train_index] y_tr = y_train.iloc[train_index] X_va = X_train.iloc[test_index] y_va = y_train.iloc[test_index] model = RandomForestRegressor(max_depth=15,n_estimators=100, random_state=777) model.fit(X_tr,y_tr)#ここで、訓練データを学習 pred_val_y = model.predict(X_va)#ここで、検証データを予測 pred_tra_y = model.predict(X_tr) pred_validation = np.append(pred_validation,pred_val_y) ans_validation = np.append(ans_validation,y_va) pred_train = np.append(pred_train,pred_tra_y) ans_train = np.append(ans_train,y_tr) val_rmse = rmse(pred_validation,ans_validation)#検証データの平均RMSEを算出 tra_rmse = rmse(pred_train,ans_train) print('(Cross validation)Train RMSE: {}, Valid RMSE: {}'.format(round(tra_rmse,3),round(val_rmse,3))) #プロットを描く plot(pred_validation,ans_validation)>(Cross validation)Train RMSE: 0.105, Valid RMSE: 0.223(出力)
ハイパーパラメータチューニング(精度向上のためモデアプローチ)
グリッドサーチ
本節では、そもそものモデルの性能を向上させるための手法である、ハイパーパラメータチューニングを紹介する。 今回は、最適化手法であるグリッドサーチを用いたチューニングについて実装する。 各機械学習アルゴリズムは、それぞれ固有のハイパーパラメータを持っている。これらは、学習により更新されるのではなく、人が予め決定する必要がある。 Random Forestであれば、決定木の深さ、決定木をいくつ集めるのかなどが存在する。グリッドサーチでは、チューニングしたいパラメータ全ての組み合わせに対して交差検証を行い、最も性能の高いパラメータの組み合わせ探索する。 今回は、3つのハイパーパラメータについてチューイングを行った。それぞれのパラメータは2つのずつ候補があるので、これらの全通り数は、8通りとなる。これら全ての通り数を実行するためにfor文を回している。from sklearn.ensemble import RandomForestRegressor #Ref.) https://scikit-learn.org/stable/modules/generated/sklearn.ensemble.RandomForestRegressor.html #以下、調節するハイパーパラメータ 。================= #通り数を増やすと結構時間がかかるので注意 parameters= {'max_depth': [15,30], #木の深さ 'min_samples_leaf': [1,2], #枝の先の最小サンプル数 'n_estimators':[10,100], #木の数 } #=========================================== best_val_rmse = np.inf for n_estimators in parameters['n_estimators']: for max_depth in parameters['max_depth']: for min_samples_leaf in parameters['min_samples_leaf']: pred_validation = np.array([]) ans_validation = np.array([]) pred_train = np.array([]) ans_train = np.array([]) for train_index,test_index in kf.split(X_train): X_tr = X_train.iloc[train_index] y_tr = y_train.iloc[train_index] X_va = X_train.iloc[test_index] y_va = y_train.iloc[test_index] model = RandomForestRegressor( max_depth=max_depth, min_samples_leaf=min_samples_leaf, n_estimators=n_estimators, n_jobs=-1, random_state=777) model.fit(X_tr,y_tr) pred_val_y = model.predict(X_va) pred_tra_y = model.predict(X_tr) pred_validation = np.append(pred_validation,pred_val_y) ans_validation = np.append(ans_validation,y_va) pred_train = np.append(pred_train,pred_tra_y) ans_train = np.append(ans_train,y_tr) val_rmse = rmse(pred_validation,ans_validation) tra_rmse = rmse(pred_train,ans_train) if val_rmse < best_val_rmse: #検証精度が改善した場合、以下の変数を上書きする。 best_val_rmse = val_rmse best_tra_rmse = tra_rmse best_predict_val = pred_validation best_predict_val_ans = ans_validation best_params={ 'max_depth':max_depth, 'min_samples_leaf':min_samples_leaf, 'n_estimators':n_estimators, 'random_state':777 'n_jobs':-1 } print('max_depth: {}, min_samples_leaf:{}, n_estimators:{}, train RMSE: {}, valid RMSE: {}'.format(max_depth,min_samples_leaf,n_estimators,round(tra_rmse,3),round(val_rmse,3))) print('Best params:') print('max_depth: {}, min_samples_leaf:{}, n_estimators:{}, train RMSE: {}, valid RMSE: {}'.format(best_params['max_depth'],best_params['min_samples_leaf'],best_params['n_estimators'],round(best_tra_rmse,3),round(best_val_rmse,3))) #診断プロットを描く plot(best_predict_val_ans,best_predict_val)>(出力) max_depth: 15, min_samples_leaf:1, n_estimators:10, train RMSE: 0.107, valid RMSE: 0.234
max_depth: 15, min_samples_leaf:2, n_estimators:10, train RMSE: 0.123, valid RMSE: 0.233
max_depth: 30, min_samples_leaf:1, n_estimators:10, train RMSE: 0.099, valid RMSE: 0.231
max_depth: 30, min_samples_leaf:2, n_estimators:10, train RMSE: 0.118, valid RMSE: 0.232
max_depth: 15, min_samples_leaf:1, n_estimators:100, train RMSE: 0.091, valid RMSE: 0.223
max_depth: 15, min_samples_leaf:2, n_estimators:100, train RMSE: 0.11, valid RMSE: 0.225
max_depth: 30, min_samples_leaf:1, n_estimators:100, train RMSE: 0.082, valid RMSE: 0.221
max_depth: 30, min_samples_leaf:2, n_estimators:100, train RMSE: 0.105, valid RMSE: 0.224
Best params:
max_depth: 30, min_samples_leaf:1, n_estimators:100, train RMSE: 0.082, valid RMSE: 0.221
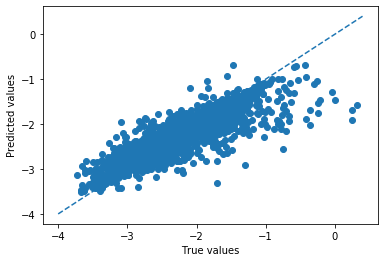
このグリッドサーチの結果から、
”max_depth: 30, min_samples_leaf:1, n_estimators:100”がベストなハイパーパラメータの組み合わせであることがわかる。 今回はチューニング方法としてグリッドサーチを用いたが、最適化する際にランダムサーチやベイズ最適化を用いる場合もある。パラメータの組み合わせ数が多すぎる場合は、これらの手法で最適化した方が良いと言えるだろう。
テストデータの予測
先ほどのパラメータを用いて、テストデータ(最終評価用データ)についての予測を行う。model = RandomForestRegressor(**best_params) #最も良かったハイパーパラメータをモデルにセットする model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) plot(y_test,y_test_pred)>RMSE(train):0.079 RMSE(test):0.212(出力)
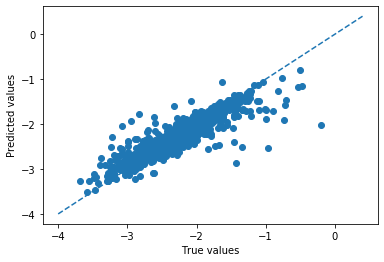
テストデータに対する予測精度が、検証精度と大きく変わらないことから過学習は生じていないことが確認できる。 また、この時の予測精度が0.2 eV/atom程度であることがわかった。第一原理計算による精度は0.01eV / atom程度であるため、それと比較するとかなり劣っていることがわかる。しかし、組成情報だけを用いて一瞬で予測が可能であることを考えると、ある程度有用に活用できるかもしれないと考えることができる。
まとめ
適切なモデル検証を行い、ハイパーパラメータチューニングをすることでより良いモデルを作成していくことが可能である。マテリアルズ・インフォマティクスの場合は、このk分割交差検証法だけを習得していれば、かなり多くの場面で役立つと思われる。マテリアルズインフォマティクスのための、機械学習の基礎(教師あり学習・回帰)
本記事の概要
この記事では、以下について解説していく。
本記事ではマテリアルズインフォマティクスで最もよく使用される教師あり学習の具体的手法について紹介する。
この記事を読むことで、機械学習モデル構築の基本を理解し、正しく実行できることを目指す。PythonやPandas、Numpyの使い方については、今回の記事では解説しない。 また、この記事のコードをテンプレートのように使い回すことも可能である。
本記事では、Materials ProjectからLi-O系化合物の生成エネルギーを取得し(取得方法については、解説していない)、このデータを用いて回帰予測を行っている。今回は、Jupyter notebookを使用してコードを書いている。
目次:
機械学習の概要
機械学習とは
おそらく、この記事を読む人は機械学習とはなんなのかわかっている人が多いと思うが、念のため説明する。
機械学習(machine learning)は、何かしらの目的を達成するための知識や行動を、データを読み込ませることで機械に獲得させるための技術である。機械学習は大きく、教師あり学習(supervised learning)、教師なし学習(unsupervised learning)、強化学習(reinforcement learning)に分けられる。本記事では教師学習についてのみ解説する。
機械に読み込ませて知識や行動を獲得させるために使うデータのことを訓練データと言う。 教師あり学習と教師なし学習の違いは、訓練データに、目的変数や説明変数(後述)があるかどうかである。もっと簡単に言うと、正解のデータがあってそれを与えるのが教師あり学習、そうでないのが教師なし学習となる。現状マテリアルズ・インフォマティクスの分野では、この教師あり学習が最もよく使用されている。
教師あり学習の手法
教師あり学習は、さらに2種類の予測が可能である。
- 回帰予測:数値を予測する
例)バンドギャップの値を予測、生成エネルギーの予測、硬度の予測など
- 分類予測:事前に定義されたラベルに分類予測する
例)絶縁体 or 導体の分類予測、硬度が高い or 低いの分類予測など
これらの予測に対して、教師あり学習には多くの手法がある。しかし、どのモデルを使用するかは、何を目指してそのモデルを構築しているのかで異なってくる。
解釈性(なぜそのような結果になったのか)を重視したい場合には、線形回帰(重回帰・Ridge回帰・Lasso回帰)や決定木など比較的簡単な予測モデルを使用することが多い。一方、高い予測精度を目指している場合には、決定木のアンサンブル(Random Forest)や深層学習を用いることが多い。そして、それらの予測精度を比較することで予測モデルを決定する必要がある。
使用するライブラリのインポート
本記事では、Scikit-learnと言う機械学習ライブラリを使用する。
Scikit-learnの公式ドキュメントには、詳細な使い方などが紹介されているので参考にすると良い。
公式ドキュメント:
scikit-learn.org
では、使用するライブラリをインポートする。
# データ加工・処理・分析ライブラリ import numpy as np import pandas as pd # 可視化ライブラリ import matplotlib.pyplot as plt %matplotlib inline #Jupyter notebook用 # 予測モデルの保存をするためのライブラリ import pickle
データの読み込み
ここでは、csv形式(コンマ区切り)のファイルを読み込んで表示する。
#Li-O系の化合物データに関する記述子、目的変数が入っているファイル名 filename='./Li_O_atom2vec_ave_var_with_form_ene_per_atom.csv' df = pd.read_csv(filename,index_col=0) df.head()

今回使用するデータは、Materials Project中に登録されている。Li - O系の組成物(約6000件)である。
データの一番先頭列には、Materials Projectにて各化合物に割り当てられているMaterial IDと組成を含んだ情報が示されている。
その後40列には、Li - O系の化合物に関する組成情報から作成した様々な組成記述子を載せている(簡単な自作組成記述子となっている)。 化合物中に存在する元素に関する特性を化合物全体の平均値、分散、最大値、最小値、最大差を取っている。
最後に、Materials Projectに登録されている物性であるFormation energy per atomが入っている。
一つの組成に対して複数の構造(多形)がある場合は、最も安定な構造(分解エネルギーが最小)となる化合物のFormation energy per atomを取得している。
予測モデルを作成・評価するための準備
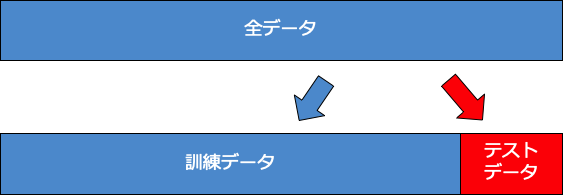
訓練データ・テストデータの作成
今回は、保留法を用いてデータの分割を行う。保留法のイメージを下に載せておく。
保留法は、単純にいテストデータを訓練データとテストデータに分割する方法である。この際のデータの選択のされ方はランダムになっている。
X = df.drop(['mp-id','formation_energy_per_atom'],axis=1) y = df['formation_energy_per_atom'] #データの分割 from sklearn.model_selection import train_test_split X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=777)
予測精度確認のための関数を事前に定義(RMSE,MAE,グラフ描画)
from sklearn.metrics import mean_squared_error #RMSEを算出するための関数を定義 def rmse(y_true,y_pred): return np.sqrt(mean_squared_error(y_true,y_pred)) #MAEを算出するための関数を定義(今回は使用しないがテンプレートとして残す) def mae(y_true,y_pred): return np.abs(y_true-y_pred).mean() #予測値と真値を比較するためのグラフを作成する関数を定義 def plot(y_true,y_pred): xx = np.arange(-4.0, 0.5, 0.1) plt.plot(xx,xx,linestyle='--') plt.scatter(y_true,y_pred) plt.xlabel('True values') plt.ylabel('Predicted values') plt.show()
予測モデルの作成・評価
重回帰
重回帰は最も簡単な回帰手法だろう。
重回帰では各記述子の係数を、予測値と目的変数の二乗誤差が最小になるように推定される。 これ以上の説明はいらないと思うので割愛する。
from sklearn.linear_model import LinearRegression #https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.LinearRegression.html model = LinearRegression()# 重回帰クラスの初期化 model.fit(X_train, y_train)#学習 y_train_pred = model.predict(X_train)# 訓練データに対する予測 y_test_pred = model.predict(X_test)# テストデータに対する予測 train_rmse = rmse(y_train,y_train_pred)# 訓練データに対するRMSE test_rmse = rmse(y_test,y_test_pred)# テストデータに対するRMSE print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse))# RMSEを表示 # 予測式の保存を行う。保存することで後からこの式を使用して予測することができる。 output_name='LinearRegression.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred) #テストデータに対する予測結果と、テストデータの真の値を比較するためのプロット
>RMSE(train):0.337 RMSE(test):0.327 (出力)

ある程度は、回帰予測ができていることが確認できる。図中の点線はy=xの直線を示しており、この線上にプロットが近づくほど、より正確な予測ができていることになる。
RIdge回帰
重回帰では、予測値と目的変数の誤差が最小化されるように学習が進んでいく。 一方、Ridge回帰では、回帰係数の二乗和も誤差に追加することで回帰係数が小さくなるように設計されている。
一般に回帰係数が大きいモデルでは、インプットの少しの動きで予測値が大きく変動するようになってしまう。 このように入力値に非常に敏感なモデルは、訓練データに対しては、非常に良い精度を示すが、未知のデータに対してはうまく当てはまらない場合があり、過学習が起きてしまうリスクがあると言える。 そこで、回帰係数の二乗和も誤差に追加することで、過学習防止を狙っているモデルがRidgeである。 一方、回帰係数の絶対和を誤差に追加するモデルは、Lassoと呼ばれる。
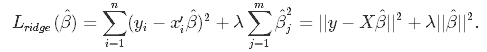
from sklearn.linear_model import Ridge #https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.Ridge.html model = Ridge(alpha=1.0) model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='Ridge.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):0.342 RMSE(test):0.335 (出力)

精度向上のためのアプローチ(スケーリング)
ここで予測精度を上げるためのアプローチの1つであるスケーリングについて紹介する。
このモデルでは、様々な記述子を使用しているが、それぞれの単位や大きさは異なっている。よって、このままだとモデルの学習が値の大きな変数に引っ張られ、値の小さな変数の影響が小さくなる懸念がある。
そこで、それを防ぐため、記述子の標準化を行う(スケーリングの方法はいくつか存在しているが、最もよく使用されているのは標準化だと思う)
標準化とはスケーリングの一種で、データの各値から変数列の平均を引き、標準偏差で割る。こうすることで変数間の単位が消え、数値の大小と意味するところが合致します。データを標準化するにはStandardScalerを使用する。
このスケーリングは、基本的に決定木系のモデル以外に関してはうまく作動するため、基本的に標準化を行った記述子を使用した方が精度が上がりやすい。
ただし、元々規格化が行われている記述子に対してさらにスケーリングを行うと精度低下を招いてしまう場合もあるので、その場合は注意が必要。
スケーリング後のRidge回帰の結果
# スケーリング(標準化) from sklearn.preprocessing import StandardScaler sc = StandardScaler() sc.fit(X_train) X_train_std = sc.transform(X_train) X_test_std = sc.transform(X_test) model = Ridge(alpha=1.0) model.fit(X_train_std, y_train) y_train_pred = model.predict(X_train_std) y_test_pred = model.predict(X_test_std) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='Ridge_std.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
> RMSE(train):0.345 RMSE(test):0.340(出力)

今回の場合は、残念ながら精度改善は見られなかった。必ずしもスケーリングによって精度が改善されるわけではないが、試す価値は大いにある。
Lasso回帰
先ほど説明したRidgeと似ているLasso回帰である。
回帰係数の絶対和を誤差に追加する事で、回帰係数が小さくなるように設計されている。
from sklearn.linear_model import Lasso #https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.Lasso.html # 重回帰クラスの初期化と学習 model = Lasso(alpha=0.0001) model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) # 決定係数を表示 print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='Lasso.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):0.350 RMSE(test):0.346(出力)

決定木回帰
決定木によるモデルは、各記述子に対して条件分岐を繰り返して予測を行う方法である。Yes or No問題を各サンプルに対して問うようなイメージである。
例えば、温度と湿度のデータから、暑く感じられるか、そうではないかといったことを分類予測する場合、以下のような決定木を作成するようにな(参考)。
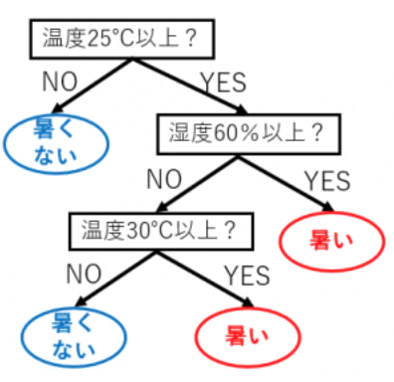
from sklearn.tree import DecisionTreeRegressor model = DecisionTreeRegressor(max_depth=15, random_state=777) model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='DecisionTreeRegression.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):0.062 RMSE(test):0.288 (出力)

RandomForest回帰
Randon Forestは、上記の決定木モデルを複数個(今回の場合は500個)作り、それらの多数決(予測値の平均値)を取って最終的な予測モデルを作成するモデルである。決定木一つだけでは、実際の予測ではそこまで高い予測精度が出ないことが多い。しかし、これら決定木を複数個足し合わせることで非常に精度の高い予測モデルを作成することができる。まさに、「三人寄れば文殊の知恵」である。実際、マテリアルズ・インフォマティクスの分野でも非常に多くの適応事例が存在している。 またハイパーパラメータチューニイングも容易であることも一つの特徴である。
from sklearn.ensemble import RandomForestRegressor model = RandomForestRegressor(max_depth=15,n_estimators=500, random_state=777) model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='RandomForestRegression.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):0.088 RMSE(test):0.211(出力)

決定木のアンサンブルであるRandomForestは、今までの予測モデルと比較するとかなり精度が高いことが確認できる。
サポートベクトル回帰
サポートベクター回帰は、回帰予測の線を一本のせんで作成するのではなく、マージン(余白)を持った線で引く手法である。概要は、以下の画像に示す。
大規模な学習データに対しては、非常に時間がかかってしまうため、中規模以下のデータに対して適応することが多い。

from sklearn.svm import LinearSVR #https://scikit-learn.org/stable/modules/generated/sklearn.svm.LinearSVR.html model = LinearSVR() model.fit(X_train, y_train) y_train_pred = model.predict(X_train) y_test_pred = model.predict(X_test) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='LinearSVR.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):3.447 RMSE(test):4.236(出力)

サポートベクトルマシンの場合、スケーリングを行った方が精度が改善する場合がある。 スケーリングを行って再度学習・予測してみる。
model = LinearSVR() model.fit(X_train_std, y_train) y_train_pred = model.predict(X_train_std) y_test_pred = model.predict(X_test_std) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='LinearSVR_std.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):0.352 RMSE(test):0.339(出力)

今回の場合は、 スケーリングにより明らかに精度が向上していることがわかる。
カーネルサポートベクトル回帰
元々の空間では、複雑だったデータ分布が高次元(カーネルより作り出された空間)においては単純な分布になる場合がある。 KernelSVRでは、データに対してカーネルを適応し、高次元空間に射影する。 その後、一つ前のセクションで行ったSVRにより回帰を行う。 カーネルとしては、しばしばRBFカーネルが使用されることが多い。カーネルによる高次元化の例を以下に示す。(参考)

from sklearn.svm import SVR # 決定木クラスの初期化と学習 model = SVR(kernel='rbf') model.fit(X_train_std, y_train) # スケーリング後の記述子を学習する y_train_pred = model.predict(X_train_std) y_test_pred = model.predict(X_test_std) train_rmse = rmse(y_train,y_train_pred) test_rmse = rmse(y_test,y_test_pred) print('RMSE(train):{:.3f} RMSE(test):{:.3f}'.format(train_rmse,test_rmse)) output_name='KernelSVR_std.pickle' with open(output_name,mode='wb') as fp:#保存するファイルを準備している。 pickle.dump(model,fp)#ここで、予測式を保存している plot(y_test,y_test_pred)
>RMSE(train):0.211 RMSE(test):0.230(出力)
